
To quantitatively analyze the nature of selection acting on these genes, we employed the McDonald-Kreitman test1 to compare the ratio of synonymous and non-synonymous substitutions between the genomes of humans and chimpanzees. In the McDonald-Kreitman test, alpha and the neutralizing index (NI) help quantify the relative strength and direction of natural selection on a gene, where a lower alpha value (higher NI) indicates higher purifying selection1.
The McDonald-Kreitman test is a robust method in molecular evolution used to detect natural selection at the molecular level. Proposed by John McDonald and Martin Kreitman in 1991, this test compares the ratio of non-synonymous (amino acid-altering) to synonymous (silent) polymorphisms within a species to the ratio of non-synonymous to synonymous fixed differences between species. The underlying premise is that synonymous changes, which do not alter protein function, are generally neutral, while non-synonymous changes can be subject to selective pressures. In the absence of selection, the ratio of non-synonymous to synonymous polymorphisms within a species is expected to be similar to the ratio of non-synonymous to synonymous fixed differences between species. Deviations from this expectation suggest the action of natural selection. Specifically, an excess of non-synonymous fixed differences indicates positive selection, while an excess of non-synonymous polymorphisms suggests balancing selection. The McDonald-Kreitman test is particularly powerful because it accounts for both within-species variation and between-species divergence, providing a comprehensive view of the evolutionary forces shaping genetic sequences. This test has become a cornerstone in the study of adaptive evolution and the detection of selective pressures on genes.
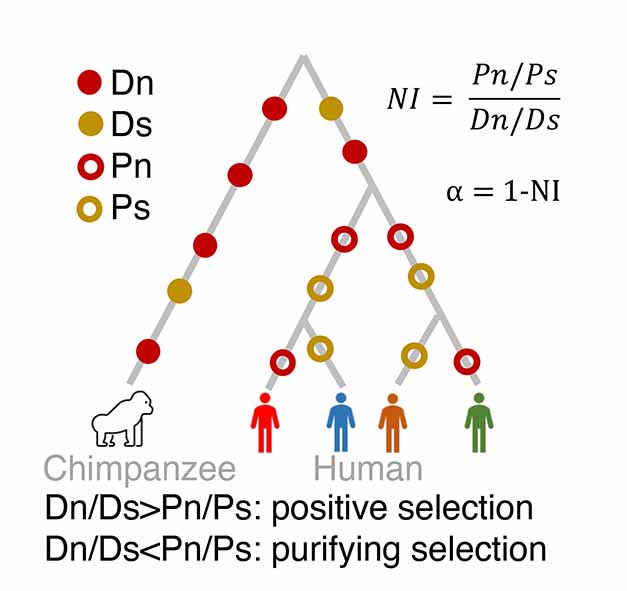
The schematic illustrates the McDonald-Kreitman test, used to estimate natural selection and infer types of selection (positive, purifying, or neutral) by analyzing patterns of synonymous and nonsynonymous substitutions between closely related species. A hypothetical tree representing the divergence between chimpanzee and human, as well as polymorphism among human populations. The filled circles represent fixed differences between chimpanzee and human (divergence). The open circles represent changes that occur among human populations (polymorphism). Synonymous and non-synonymous substitutions are represented by yellow and red circles, respectively. The McDonald-Kreitman test compares the ratio of non-synonymous divergence (Dn; red filled circle) to synonymous divergence (Ds; yellow filled circle) (Dn/Ds) with the ratio of non-synonymous polymorphic changes (Pn; red open circle) to synonymous polymorphic changes (Ps; yellow open circle) (Pn/Ps). Positive selection can be detected when the ratio of non-synonymous divergence to synonymous divergence (Dn/Ds) is greater than the ratio of non-synonymous polymorphic changes to synonymous polymorphic changes (Pn/Ps).
In this study, the natural selection of protein-coding genes was estimated using the McDonald–Kreitman (MK) test1 by inferring the proportion of positive selection (α)2 and Neutrality index (NI)3. The MK test compares the ratio of nonsynonymous to synonymous substitutions within a species (polymorphism) to the ratio of nonsynonymous to synonymous fixed differences between this species and its closely related species (divergence). Here, we use chimpanzee as the outgroup to calculate the divergence. NI and α values were determined as follows:
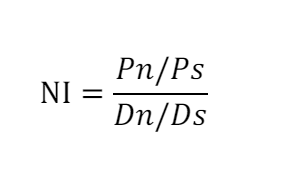
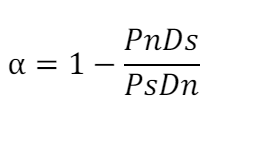
where Pn is the number of nonsynonymous variants within the species, Ps is the number of synonymous variants within the species; Dn and Ds represent the numbers of nonsynonymous and synonymous variants between species, respectively. An NI value greater than 1 indicates purifying selection in effect, while an NI value less than 1 indicates positive selection. A positive α value suggests positive selection, whereas α < 0 suggests negative selection. To calculate NI and α values, we retrieved human variants from the 1000 Genomes Project Phase 3 GRCh38, which included 2548 samples. The human-chimpanzee alignment and variant calling data were retrieved from Prado-Martinez J, et al.4, which included 25 samples. Since the genome build of the chimpanzee variant data was mapped to hg18, liftover to human GRCh38 was performed using Picard (https://broadinstitute.github.io/picard/). The VCF files of human and chimpanzee were merged and submitted to the R package ‘popgenome’ for the estimation of NI and α values for each protein-coding gene using the function ‘MKT’.
References:
1. McDonald, J.H. & Kreitman, M. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652-654 (1991).
2. Smith, N.G. & Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 415, 1022-1024 (2002).
3. Rand, D.M. & Kann, L.M. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol 13, 735-748 (1996).
4. Prado-Martinez, J. et al. Great ape genetic diversity and population history. Nature 499, 471-475 (2013).